The Basics
Each clinical trial must go through many layers of review before it can begin. Every layer of the process is intended to protect your safety.
The U.S. government has strict guidelines and safeguards in place to keep risks as low as possible and ensure that any risks are worth the potential benefits. On top of those requirements, we add additional safety measures for trials in CF. The infographic below explains all the ways your safety is protected as a study volunteer, both before the study begins and while it is happening.
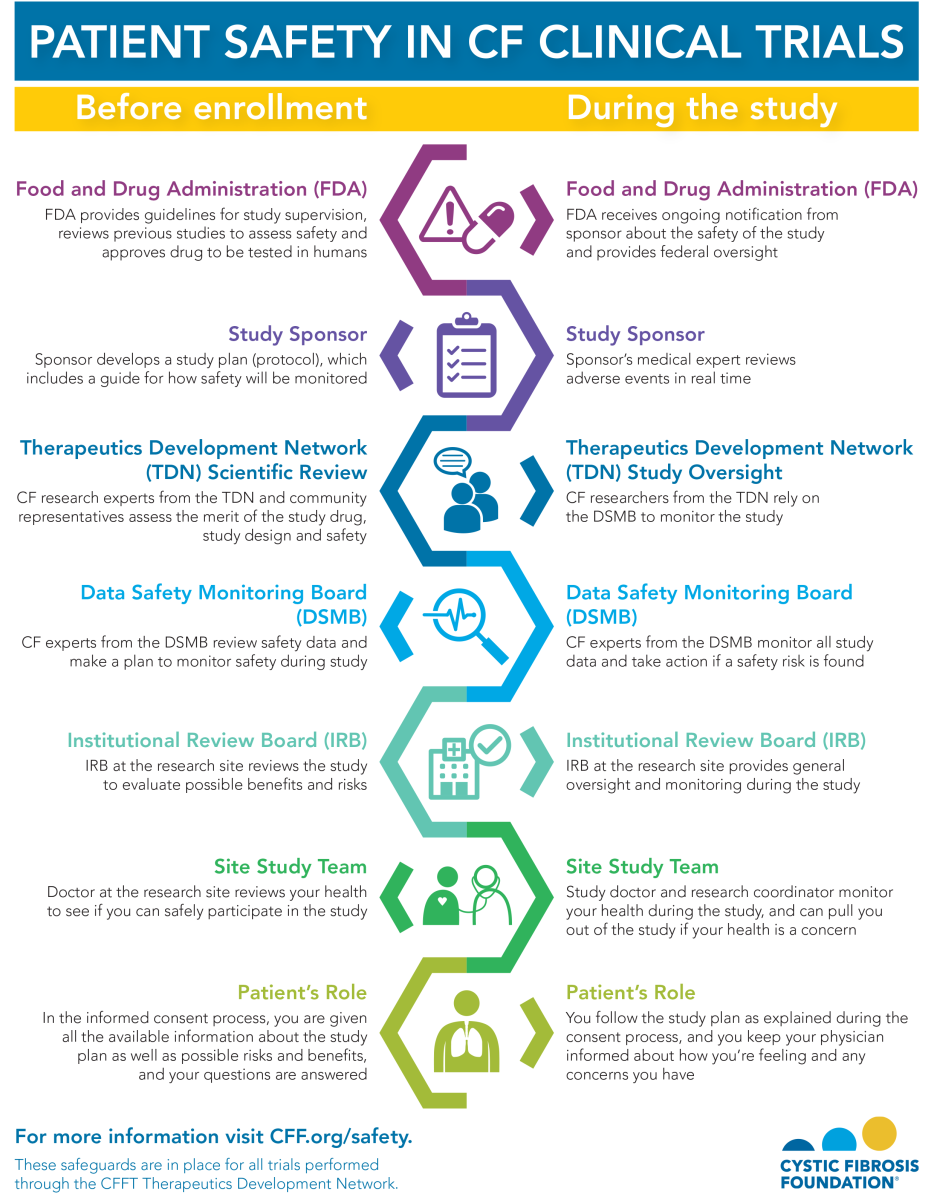
The Details: All the Ways Your Safety Is Protected
U.S. Food and Drug Administration (FDA)
Before a potential medication or therapy can be tested in people, it must be reviewed and approved for further study by the FDA. The sponsor of the study -- usually the drug company -- must submit an application for an investigational new drug. In this application, the FDA reviews the results from initial laboratory studies to make sure that the potential drug poses very few risks to people and that any risks are worth the possible benefits.
Once the FDA approves the application for an investigational new drug, it must approve all clinical trial plans (protocols) and make sure all required guidelines are being followed. These guidelines ensure that every clinical trial is safe and ethical.
Study Sponsor and Medical Monitors
The study sponsor develops the protocol, which includes a guide for how safety will be monitored. During the study, medical experts hired by the sponsor monitor adverse events in real time. Adverse events are considered negative side effects that one may experience, whether they are related to the study drug or not. For example, an increased cough during a clinical trial would be considered an adverse event, and it would be monitored throughout the study.
Therapeutics Development Network
After the FDA approves the clinical trial, the sponsor submits the trial to the Therapeutics Development Network (TDN). The TDN is a collaborative network of care centers that have expertise in conducting CF clinical trials. The CF Foundation is the primary source of funding for the TDN.
The TDN Coordinating Center, based in Seattle, conducts several reviews before approving a trial:
- A compound review is conducted when multiple drug compounds in the same treatment category (such as anti-inflammatories) are being developed for clinical trials. Independent experts review the new study drug to determine if it is likely to be more effective than other existing drugs in the same category. If it isn't, the compound will not move forward into clinical trials within the TDN.
- The Protocol Review Committee conducts a thorough review of the protocol and generates a detailed critique, as well as ratings for scientific merit, feasibility, and study design. This committee is made up of a pool of experienced CF physician researchers, research coordinators, biostatisticians, people with CF, and other specialists. In addition to maximizing participant safety, this review helps sponsors understand how their studies fit with the priorities of the CF community.
- The Clinical Research Executive Committee assesses how the protocol fits within the overall priorities of the Foundation for clinical research and assigns a numerical rating for strategic fit. This committee includes CF Foundation executives and senior CF researchers.
Data Safety Monitoring Board
All clinical trials must have a data safety monitoring board (DSMB), an independent committee of experts in clinical care, clinical and basic science research, bioethics, and biostatistics, as well as people with CF. The primary responsibility of this board is to protect the safety and welfare of people who participate in Foundation-sanctioned clinical trials and to ensure the accuracy and consistency of those trials.
“My patients are my heroes … And when they volunteer to do this research, they need to be protected and that's what we do.” — Wayne Morgan, M.D., chairman of the Data Safety Monitoring Board funded by the Foundation
The DSMB takes many steps to ensure trials are safe, including:
- Reviewing the protocol before the study begins to make sure it includes an appropriate plan to monitor participants' safety
- Monitoring the study in real time to pick up early signs of potential side effects
- If necessary, suggesting changes or even stopping the trial to protect participants' safety
Although both the TDN and DSMB are funded by the Foundation, they are separate from each other and have separate review processes. The protocol reviews complement each other and are done in parallel. DSMB members are totally independent experts in CF and are not involved in any way with the trial, the sponsor, or its participants.
Wayne Morgan, M.D., chairman of the Data Safety Monitoring Board funded by the Foundation, explains the importance of patient safety, how the Data Safety Monitoring Board protects patients, the patient’s role before joining a study, and what makes the Data Safety Monitoring Board exceptional.
Institutional Review Board
After a clinical trial has been reviewed by the FDA, the TDN, and the DSMB, the next level of review happens at the local level. Every care center that is conducting the trial has an institutional review board (IRB), an independent committee that reviews the entire study proposal.
The IRB review includes:
- Determining whether the benefits of the trial are greater than the risks
- Ensuring that volunteers will be monitored closely for safety
- Verifying that volunteers have been given information that is easy to understand and allows them to make an informed decision about participation
IRBs will only approve research that deals with medically important questions in a scientific and responsible way. The IRB at each study location will continue to monitor study conduct throughout the trial and is notified of any adverse events.
Site Principal Investigator
The study site principal investigator (PI), usually a CF doctor, is the leader of the research team. They are responsible for the oversight of a study at a particular site. The most important part of the PI's responsibility is to monitor the safety of the study participants at their site.
Before enrollment, the PI reviews your health to see if you can safely participate in the study. During the study, the PI reviews all safety measurements including lung function, blood work, and any changes in symptoms or overall health.
The PI's first priority is always the participant's safety. If at any time the PI feels the study is not in your best interest, the PI has not only the right -- but also the duty -- to remove you from the study.
“Your care team isn't going to intentionally put you at risk or weigh the odds when it comes to your health -- it always comes first. I think trusting that is important.” -- Jodi Marquez, an adult with CF, who has participated in two clinical trials
Your Role as a Study Participant
In the informed consent process, you are given all the available information about the study, including the time commitment, procedures, and possible risks and benefits. It is important for you to ask as many questions as needed so that you can feel comfortable about participating.
During the study, you are responsible for following the study plan as explained to you during the informed consent process. It's important to keep your doctor informed about how you're feeling and any concerns you may have. Before enrolling in a study, ask who you should contact with concerns: your regular care team, the research team, or both.
Remember, your safety is the priority. If you feel that it's not in your best interest to continue, you can drop out of a trial at any time.