Over the last four years, CFTR modulators that restore CFTR function have been approved for over 50 percent of people with cystic fibrosis. This is one of the most exciting advances to happen in CF; however, some individuals carry mutations that are not able to benefit from these medications. It is with these individuals in mind that the Cystic Fibrosis Foundation today opened this year's North American CF Conference (NACFC).
In the first plenary of the NACFC, A Cure for All: Leaving No One Behind, two longtime CF researchers made it clear that a broad range of strategies is being explored to develop treatments for everyone with CF, especially those who carry mutations that cannot benefit from ivacaftor (Kalydeco®) and lumacaftor/ivacaftor (Orkambi®).
As director of research at the Cystic Fibrosis Foundation, I have the privilege of working with the scientists who are focused on identifying new targets and developing treatments. It is gratifying to see the progress being made on all fronts, and I wanted to share some of the advances in the field highlighted by Drs. Eric Sorscher and Mitchell Drumm during the first plenary session.
First, early lab results indicate that the second-generation CFTR modulators will be more effective than ivacaftor and lumacaftor/ivacaftor and may provide options for people with CF who have only one copy of the F508del mutation, according to Dr. Sorscher, the Georgia Research Alliance Eminent Scholar and Hertz Professor in CF Research at Emory University in Atlanta. If clinical trials for these drugs are successful, nearly 95 percent of people with CF will have a drug that targets the underlying cause of CF within the next few years.

That leaves 5 percent of people with CF without any treatment that targets the underlying cause of their CF symptoms, the defective CFTR protein. But rest assured that research is being conducted to ensure that no one -- even those with the rarest mutations -- is left behind.
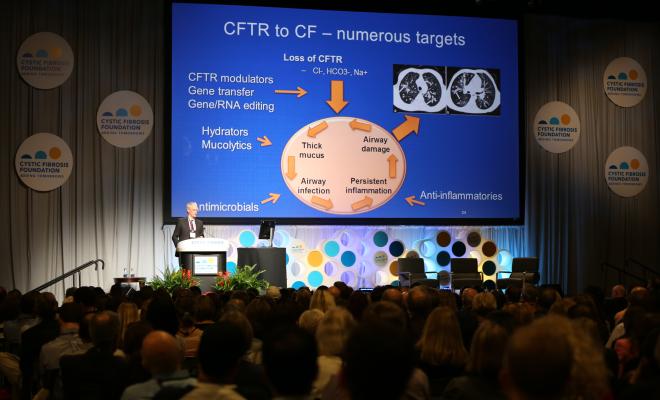
The CFTR protein is key in CF because it regulates the proper flow of fluids and salt (chloride) in and out of cells lining the lungs and other organs. Because CFTR modulators correct problems with the CFTR protein, they do not help those people who have mutations, such as nonsense mutations, in which no protein is made.
In people with CF who have nonsense mutations, a signal prematurely stops production of the CFTR protein, resulting in a shortened and nonfunctional protein. In addition, messenger RNA, which carries the genetic instructions to make protein, degrades before the protein can be made.
The CF Foundation is supporting a number of efforts to find compounds that can stabilize the messenger RNA and read through the premature stop signals so as to make adequate amounts of functional CFTR protein. We see this as just one piece in the puzzle as we strive to meet our goal of ensuring everyone with CF benefits from treatments that restore CFTR function.
As more people with CF gain access to current and future CFTR modulators at younger ages, the overall health of the CF population will improve before serious lung damage occurs. In the coming decades, we are going to see a significant change in the demographics of the CF population, Dr. Sorscher said, but right now we need to focus on developing therapies that have the potential to benefit all people with CF today. Although we hope that people with CF will need fewer symptomatic treatments over time, the development of better treatments to address the complications of the disease is still as important as ever, he said. Reducing excessive inflammation, fighting chronic infections and improving airway clearance remain top priorities.
Excessive inflammation is the main cause of lung tissue damage in people with CF. Researchers from academia and industry are investigating different approaches to reduce the body's inflammatory response and boost the natural processes that help control inflammation. CF Foundation Therapeutics (CFFT) is currently providing funding for the clinical development of five potential anti-inflammatory drugs.
Chronic infections, especially by drug-resistant bacteria, are another top concern. Researchers are testing new formulations and new uses of existing antibiotics. Clinical trials are underway for an inhaled version of the antibiotic vancomycin for the treatment of airway infections with methicillin-resistant Staphylococcus aureus (MRSA). Another clinical trial that just recently started recruiting patients is testing gallium, a drug that has already been approved by the U.S. Food and Drug Administration for intravenous use in another disease. Gallium is a molecule that has been shown to kill antibiotic-resistant strains of Pseudomonas aeruginosa in the laboratory and may serve as a new therapeutic option for patients with hard-to-treat infections.
Several drugs are also under development to thin or alter the thick, sticky mucus in the lungs of people with CF so that it can be coughed out more easily. One approach is to block the function of a sodium channel found in the lungs in order to maintain fluid within the airways and help improve airway clearance.
Our goal with all these treatments is to keep people with CF as healthy as possible until we can deliver a cure, Dr. Sorscher said. To do that, the CF Foundation is funding fundamental research into new technologies to attack the basic defect in CF on all fronts.
Dr. Drumm, a professor of pediatrics and genetics for Case Western Reserve University School of Medicine, explained that editing the CF gene holds the promise of a permanent fix for CF. He explained the potential of the CRISPR Cas9 editing technology, which has revolutionized science in recent years, but also highlighted the difficulty in bringing it from the lab to the clinic.

For example, we still don't know if one edit will work for every mutation, or whether we will need to correct each of the more than 1,700 mutations in CF. In addition, scientists still have to determine which cells are best to target in the lung, how to reach them with the gene editing tools and how many cells will need to be edited to make a clinical difference.
Many similar challenges apply to RNA therapy. RNA therapy involves either adding normal messenger RNA directly to cells or repairing the defective messenger RNA in the cell for production of a normal CFTR protein. While these therapeutic options would be an improvement over the daily regimens offered by CFTR modulators, correcting the basic defect in the DNA is still the ultimate goal to eliminate the disease altogether.
A number of significant advances have been made in the past few years that have changed the course of this disease, many of which seemed impossible not long ago. This is due to the dedication of a team of researchers, health care providers and people with CF all working together to advance a common goal. Although we have lots of work ahead, this truly is one of the most exciting times to be a part of the CF research community.